Density Functional Calculations for Amino Acids
Density Functional Calculations for Amino Acids
Within the framework of a diploma thesis [1] the structure of three
amino acids (glycine, alanine, serine) has been studied, using
density functional methods.
Utilizing the Car-Parrinello technique [2], the calculations are based
on
-
the representation of the ionic cores by
normconserving pseudopotentials (applying the
Troullier-Martins form [3] and the Kleinman-Bylander transformation
[4]),
-
a plane-wave expansion of the single-particle Kohn-Sham states,
-
the supercell concept for periodic repetition of the molecules
(using a simple cubic unit cell),
-
the local density approximation (in the
VWN parameterization [5]) or the generalized
gradient approximation (in the PW form [6]) for the
exchange-correlation energy functional,
-
a quasi-Newton method for the relaxation of the ionic positions, i.e.
the total energy minimization
(Broyden-Fletcher-Goldfarb-Shanno algorithm).
It has been demonstrated (see e.g. [7]) that for given
exchange-correlation functional the spectroscopic constants obtained
with normconserving pseudopotentials agree excellently with the
corresponding all-electron data, if (i) sufficiently conservative
cut-off radii are used and (ii) nonlinear core corrections [8] are
taken into account.
However, the inclusion of nonlinear core corrections mainly affects
dissociation energies, it is less relevant for all geometrical aspects.
Moreover, a suitable choice of the cut-off radii is straightforward.
The quality of the present results is thus only controlled by the form
of the exchange-correlation functional and two technical parameters,
-
the basis set size --
in the case of plane-wave basis sets this size is determined by the
highest energy present in the basis, the cut-off energy --
-
and the size of the supercell, which has to make sure that the
neighboring molecules do not interact.
The latter size has been chosen so that atoms belonging to different
molecules are separated by roughly 10 Bohr.
The results can be summarized as follows:
-
The structures of all low-lying conformers examined in this work are
obtained rather accurately by density functional methods.
-
The same is not true for the energetic ordering of the various states:
The subtle energy differences involved can not always be reproduced by
the LDA or GGA, indicating the limitations of these (semi)local
functionals.
-
Gradient corrections appear to be less important for these organic
molecules than for many solids.
-
The calculations demonstrate the feasibility of DFT calculations for
comparatively complex systems on present day consumer PC's.
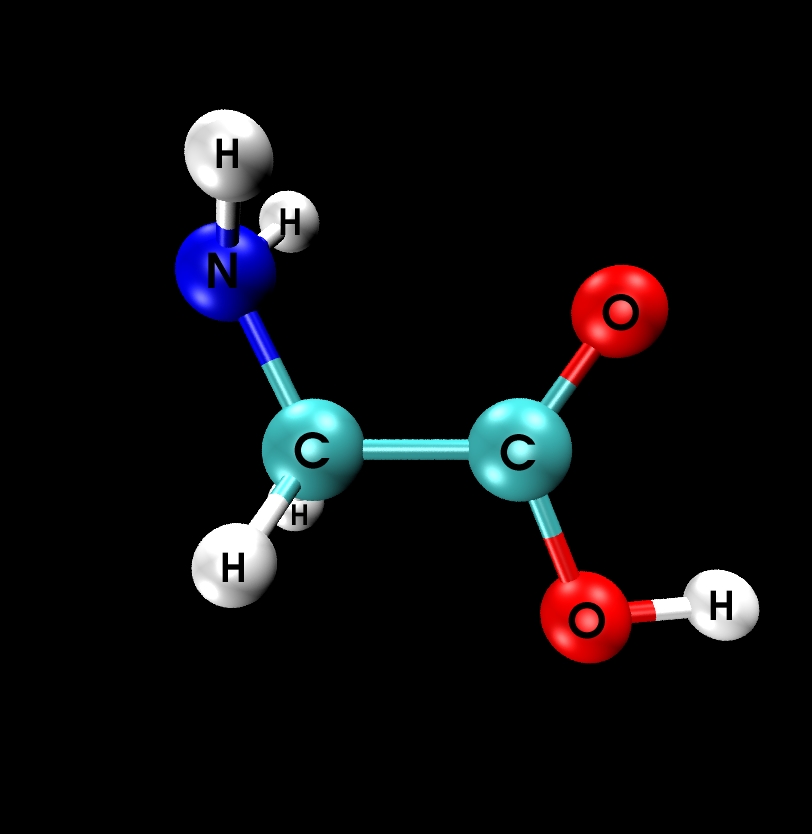
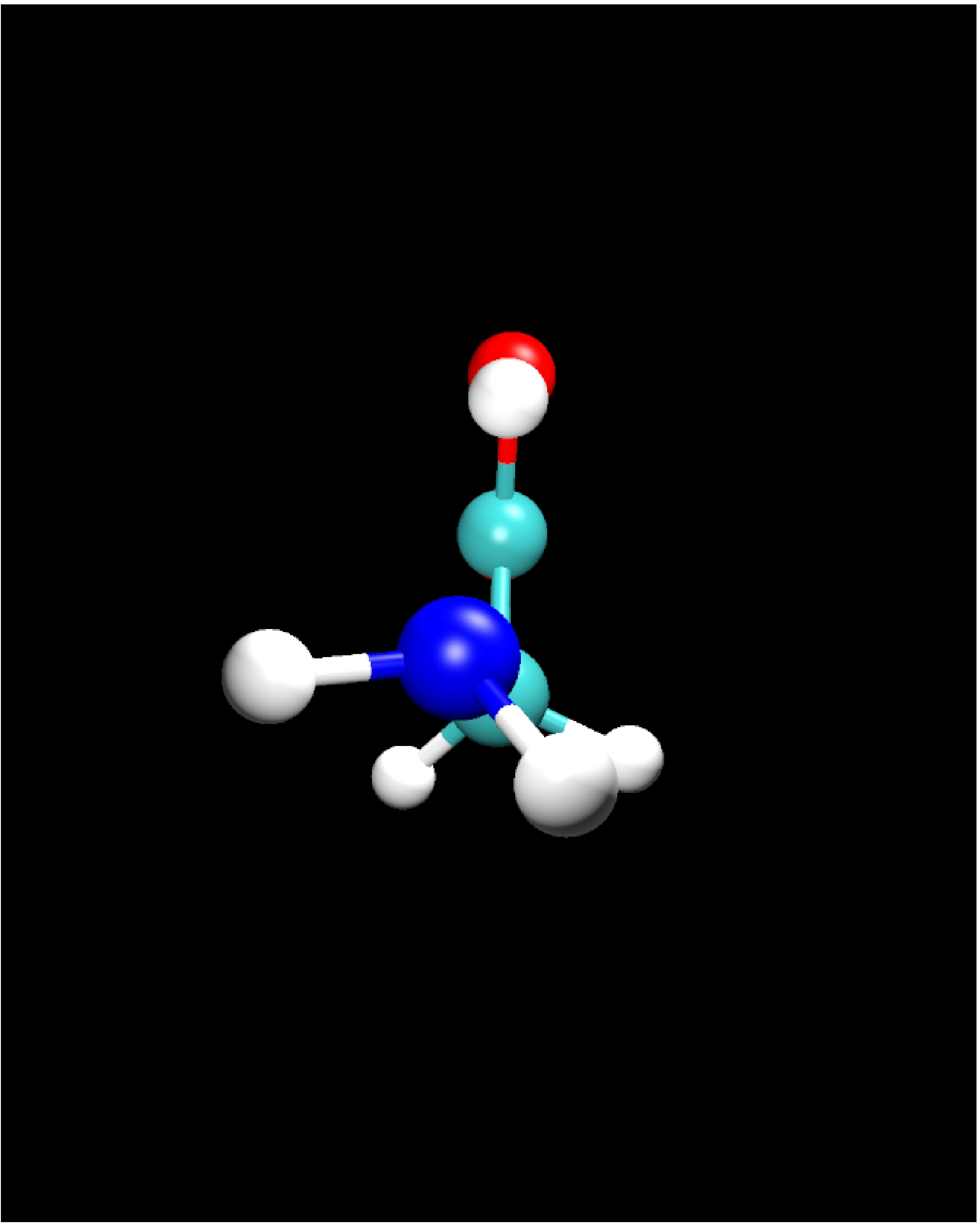
II.1) Ground state: Glycine Ip (planar)
|
Calculational parameters
|
| xc-functional |
LDA [5] |
| cut-off energy |
100 Rydberg |
| pseudopotential |
Troullier-Martins [3] |
| nonlinear core corrections |
no |
| supercell size |
23 Bohr |
|
Results
|
bond lengths
[Angstroms] |
bond angles
[degrees] |
| bond |
expt. [9] |
LDA |
angle |
expt. [9] |
LDA |
| N-H |
(1.001) |
1.020 |
H-N-H |
(110.3) |
106.5 |
| N-C |
1.467 |
1.431 |
H-N-C |
(113.3) |
111.0 |
| C-H |
(1.081) |
1.101 |
N-C-C |
112.1 |
115.9 |
| C-C |
1.526 |
1.506 |
H-C-H |
(107.0) |
104.4 |
| C-O |
1.355 |
1.332 |
C-C-O |
111.6 |
114.9 |
| C=O |
1.205 |
1.200 |
C-C=O |
125.1 |
125.7 |
| O-H |
(0.966) |
0.976 |
C-O-H |
(112.3) |
104.7 |
- experimental results in brackets are based on estimates
- reasonable agreement between LDA and experiment
- however: LDA erroneously predicts conformer IIp to be energetically
lower than the Ip state by 0.14 eV
|
|
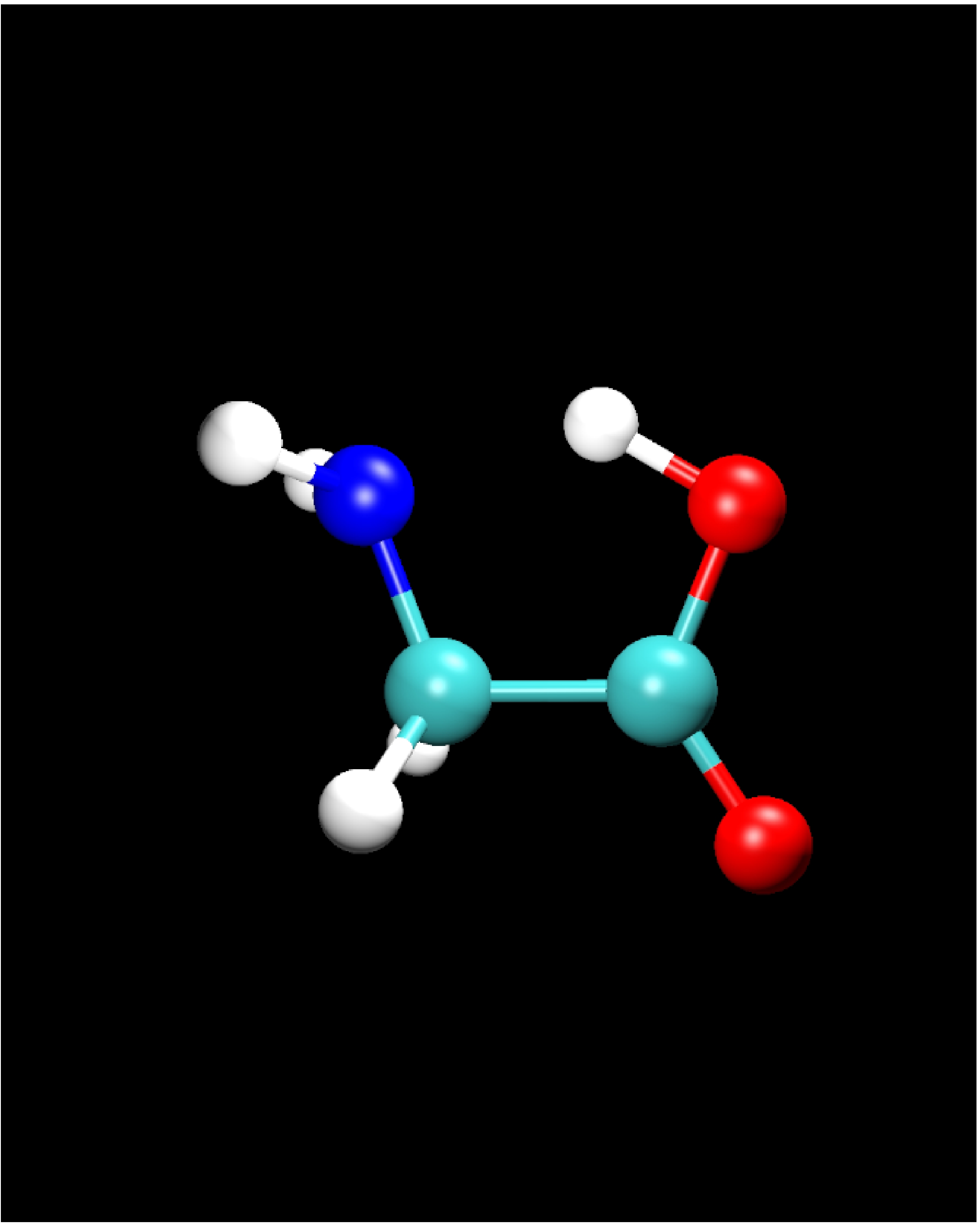
II.2) Energetically lowest conformer: Glycine IIp
|
Calculational parameters
|
| xc-functional |
LDA [5] |
| cut-off energy |
60 and 100 Rydberg |
| pseudopotential |
Troullier-Martins [3] |
| nonlinear core corrections |
no |
| supercell size |
23 Bohr |
|
Results
|
bond lengths
[Angstroms] |
bond angles
[degrees] |
| bond |
LDA
60 Ryd |
LDA
100 Ryd |
angle |
LDA
60 Ryd |
LDA
100 Ryd |
| N-H |
1.026 |
1.019 |
H-N-H |
107.9 |
108.7 |
| N-C |
1.453 |
1.449 |
H-N-C |
113.1 |
113.7 |
| C-H |
1.103 |
1.099 |
N-C-C |
109.9 |
110.4 |
| C-C |
1.518 |
1.518 |
H-C-H |
106.2 |
106.1 |
| C-O |
1.338 |
1.324 |
C-C-O |
112.2 |
112.0 |
| C=O |
1.228 |
1.203 |
C-C=O |
122.5 |
123.1 |
| O-H |
1.046 |
1.023 |
C-O-H |
100.0 |
101.8 |
-
A bond contraction is observed, when increasing the basis set
size from 60 to 100 Rydberg: The oxygen bonds are affected most
strongly.
-
Results for 80 Rydberg are almost identical with those for 100
Rydberg.
|
|
|
Calculational parameters
|
| xc-functional |
GGA [6] |
| cut-off energy |
80 Rydberg |
| pseudopotential |
Troullier-Martins [3] |
| nonlinear core corrections |
no |
| supercell size |
23 Bohr |
|
Results
|
bond lengths
[Angstroms] |
bond angles
[degrees] |
| bond |
GGA |
angle |
GGA |
| N-H |
1.013 |
H-N-H |
107.9 |
| N-C |
1.453 |
H-N-C |
112.8 |
| C-H |
1.088 |
N-C-C |
110.5 |
| C-C |
1.518 |
H-C-H |
106.4 |
| C-O |
1.344 |
C-C-O |
112.8 |
| C=O |
1.224 |
C-C=O |
122.7 |
| O-H |
1.020 |
C-O-H |
101.1 |
-
GGA contracts bonds slightly compared to LDA: Effect of gradient
corrections is less dramatic than for many inorganic molecules and
for solids.
|
|
II.3) Glycine IIn
|
Calculational parameters
|
| xc-functional |
LDA [5] |
| cut-off energy |
80 Rydberg |
| pseudopotential |
Troullier-Martins [3] |
| nonlinear core corrections |
no |
| supercell size |
23 Bohr |
|
Results
|
bond lengths
[Angstroms] |
bond angles
[degrees] |
| bond |
LDA |
angle |
LDA |
| N-H |
1.021 |
H-N-H |
108.5 |
| N-C |
1.452 |
H-N-C |
113.7 |
| C-H |
1.099 |
N-C-C |
110.3 |
| C-C |
1.518 |
H-C-H |
106.2 |
| C-O |
1.330 |
C-C-O |
112.0 |
| C=O |
1.206 |
C-C=O |
123.1 |
| O-H |
1.029 |
C-O-H |
101.3 |
|
|
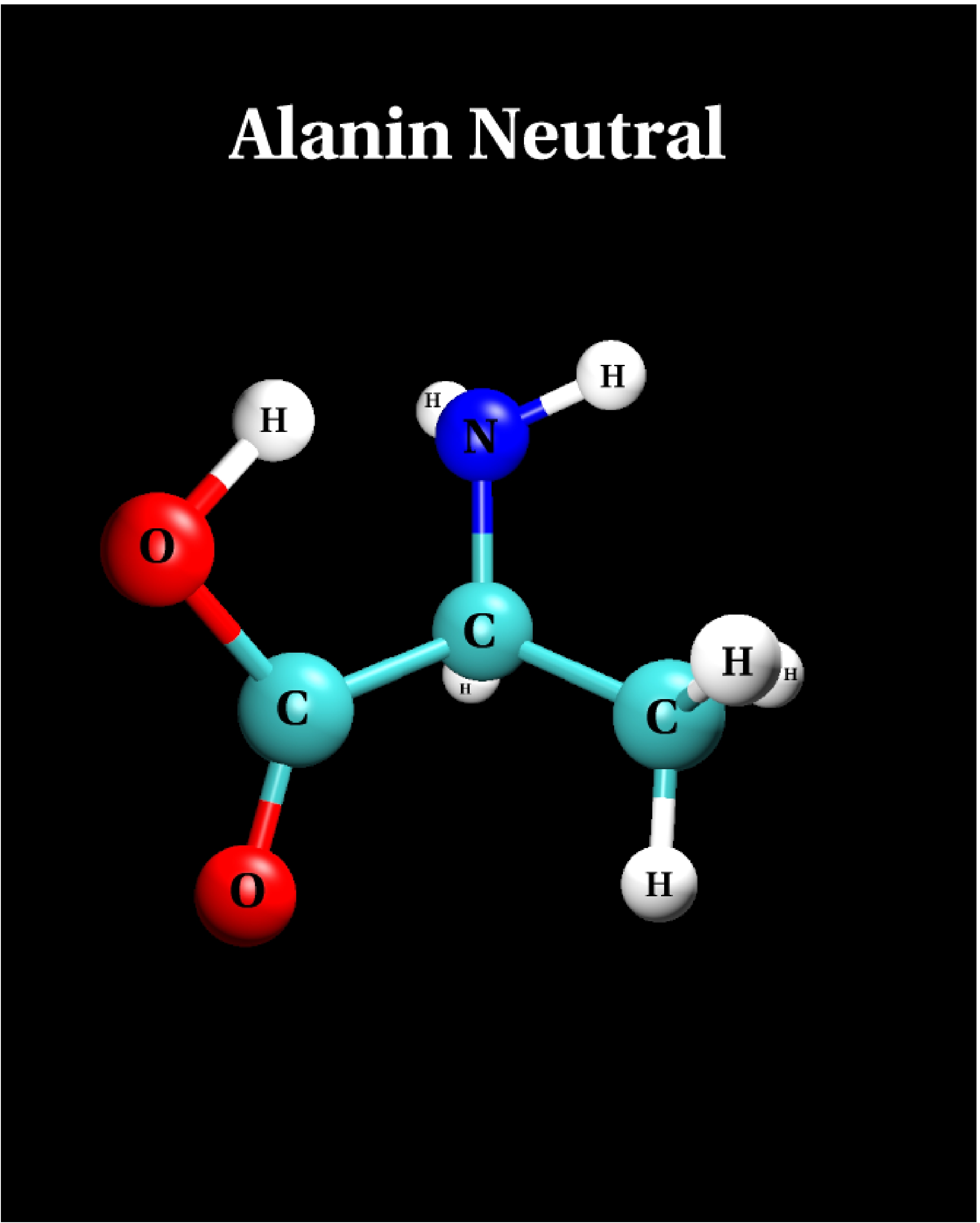
III.1) Neutral structure
|
Calculational parameters
|
| xc-functional |
LDA [5] |
| cut-off energy |
60 Rydberg |
| pseudopotential |
Troullier-Martins [3] |
| nonlinear core corrections |
no |
| supercell size |
23 Bohr |
|
Results
|
bond lengths
[Angstroms] |
bond angles
[degrees] |
| bond |
LDA |
angle |
LDA |
| N-C |
1.458 |
- |
- |
| C-C |
1.526 |
N-C-C |
108.3 |
| C-O |
1.334 |
C-C-O |
112.6 |
| C=O |
1.230 |
C-C=O |
122.3 |
-
The LDA predicts the neutral structure to be the ground state
configuration if alanine, in contrast to quantum chemical
results [10].
The LDA energy difference to the zwitterionic state is 0.94 eV.
|
|
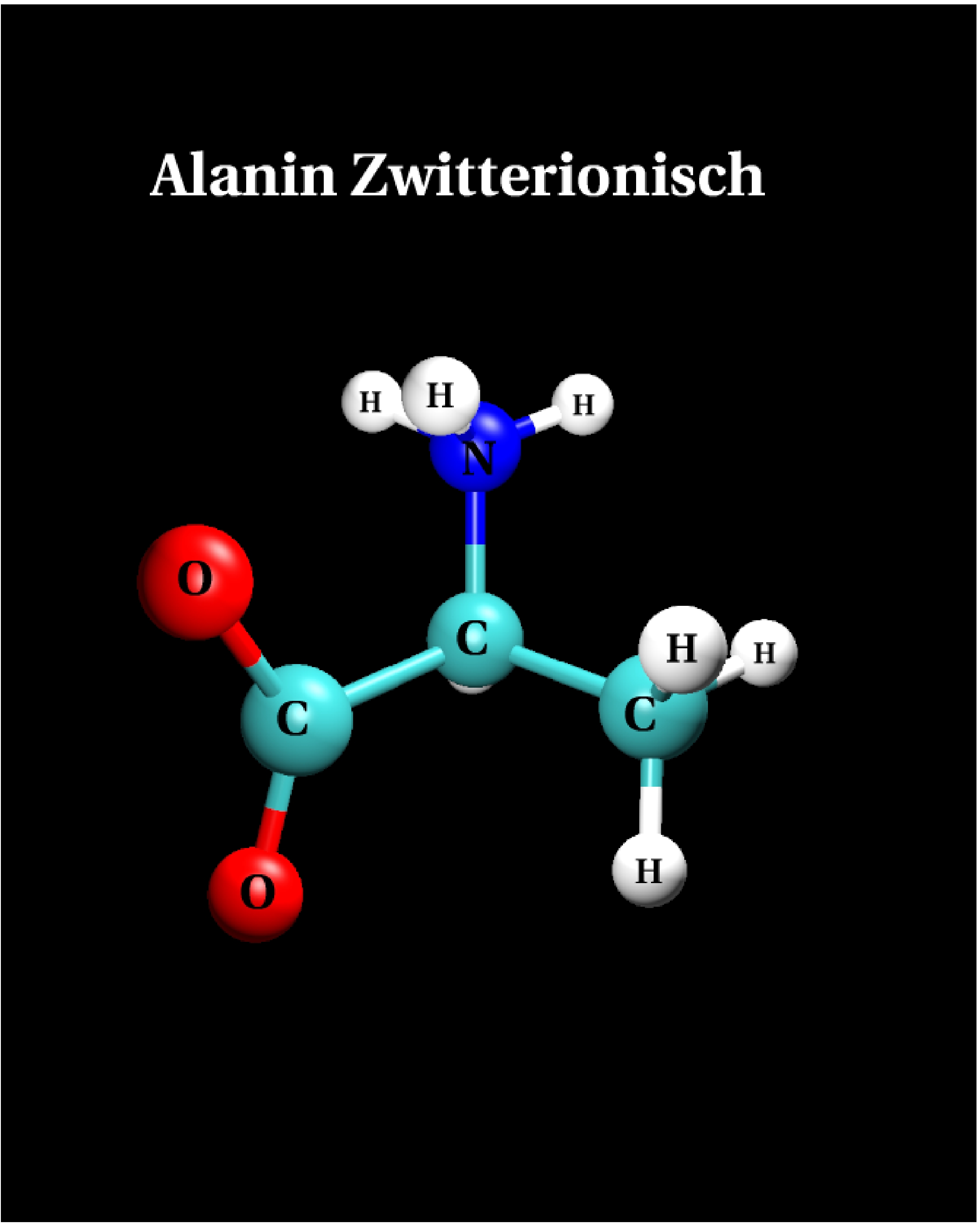
III.2) Zwitterion
|
Calculational parameters
|
| xc-functional |
LDA [5] |
| cut-off energy |
60 Rydberg |
| pseudopotential |
Troullier-Martins [3] |
| nonlinear core corrections |
no |
| supercell size |
23 Bohr |
|
Results
|
bond lengths
[Angstroms] |
bond angles
[degrees] |
| bond |
LDA |
angle |
LDA |
| N-C |
1.481 |
- |
- |
| C-C |
1.564 |
N-C-C |
106.6 |
| C-O |
1.287 |
C-C-O |
115.5 |
| C=O |
1.251 |
C-C=O |
116.6 |
|
|
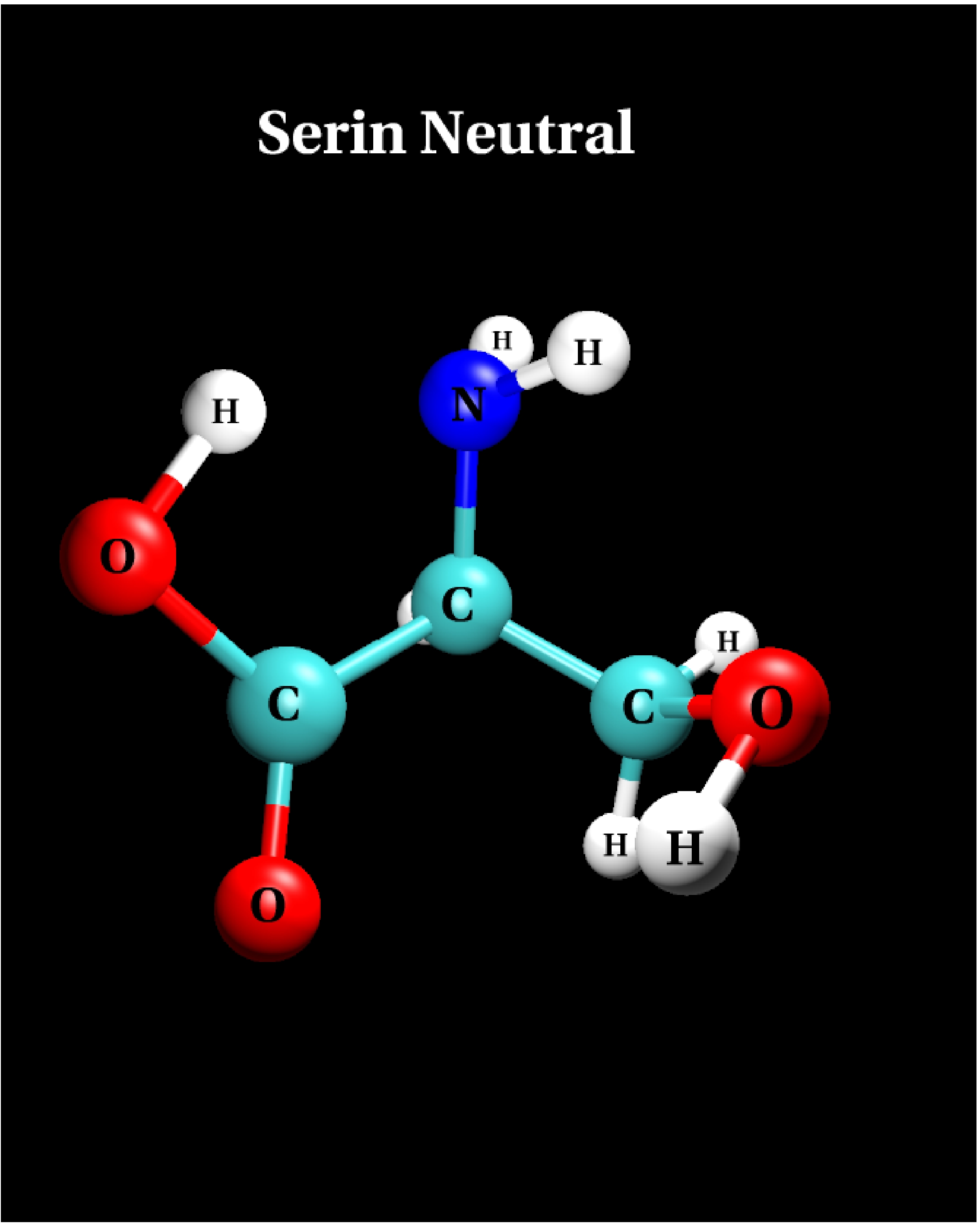
IV.1) Ground state
|
Calculational parameters
|
| xc-functional |
LDA [5] |
| cut-off energy |
60 Rydberg |
| pseudopotential |
Troullier-Martins [3] |
| nonlinear core corrections |
no |
| supercell size |
23 Bohr |
|
Results
|
bond lengths
[Angstroms] |
bond angles
[degrees] |
| bond |
LDA |
angle |
LDA |
| N-H |
1.028 |
H-N-H |
108.6 |
| N-C |
1.452 |
H-N-C |
113.2 |
| C-H |
1.107 |
N-C-C |
107.0 |
| C-C |
1.528 |
H-C-H |
109.0 |
| C-O |
1.333 |
C-C-O |
112.4 |
| C=O |
1.232 |
C-C=O |
122.3 |
| O-H |
1.054 |
C-O-H |
99.4 |
|
|
-
G. Iseri,
Diploma thesis, University of Frankfurt (2003).
-
M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias,
and J. D. Joannopoulos,
Rev. Mod. Phys. 64, 1045 (1992).
-
N. Troullier and J. L. Martins,
Phys. Rev. B 43, 1993 (1991).
-
L. Kleinman and D. M. Bylander,
Phys. Rev. Lett. 4, 1425 (1982).
-
S. H. Vosko, L. Wilk, and M. Nusair,
Can. J. Phys. 58, 1200 (1980).
-
J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson,
M. R. Pederson, D. J. Singh, and C. Fiolhais,
Phys. Rev. B 46, 6671 (1992).
-
E. Engel, A. Höck, R. N. Schmid, R. M. Dreizler, and N. Chetty,
Phys. Rev. B 64, 125111 (2001).
-
S. G. Louie, S. Froyen, and M. L. Cohen,
Phys. Rev. B 26, 1738 (1982).
-
Iijima, Tanaka, Onuma,
J. Mol. Struct. 246, 257 (1991).
-
F. R. Tortonda, J.-L. Pascual-Ahuir, E. Silla, and I. Tunon,
J. Chem. Phys. 109, 592 (1998).
Home
Top
Last modified: April 2, 2004
Disclaimer
ee